Minimal example¶
import numpy as np
import circe as ci
import scanpy as sc
import scipy as sp
Import (or create) data¶
Creating fake AnnData¶
This data doesn’t contain strong correlation between fake regions, the score will then be low.
It will still allow us to demonstrate how to use Circe. :)
atac = sc.datasets.blobs(n_centers=8, n_variables=2_000, n_observations=300, random_state=1)
atac.X = np.random.poisson(lam=0.2, size=atac.X.shape)
cell_names = [f"cell_{i}" for i in range(1, atac.shape[0]+1)]
# number of chr_start_end region names
region_names = [[f"chr{i}_{str(j)}_{str(j+399)}"
for j in range(1, 10000*400+1, 10000)]
for i in range(1, 6)]
regions_names = [item for sublist in region_names for item in sublist]
# select a random number of regions per cell, and make it 0
for i in range(atac.shape[0]):
num_regions = np.random.randint(500, 1000)
region_indices = np.random.choice(len(regions_names), num_regions, replace=False)
atac.X[i, region_indices] = 0
atac.var_names = regions_names
atac.obs_names = cell_names
1. Preprocessing the data¶
1.1. Filtering the data¶
sc.pp.filter_genes(atac, min_cells=30)
sc.pp.filter_cells(atac, min_genes=20)
Make sure to all your features (peaks) are expressed in at least 1 of the cells:
sc.pp.filter_genes(atac, min_counts=1)
1.2. Adding region position in AnnData.obs¶
Let’s first run add_region_infos that will add coordinate annotations chr, start, end as columns in atac.var slot
atac = ci.add_region_infos(atac)
atac.var.head(3)
| n_cells | n_counts | chromosome | start | end | |
|---|---|---|---|---|---|
| chr1_1_400 | 34 | 38 | chr1 | 1 | 400 |
| chr1_10001_10400 | 31 | 31 | chr1 | 10001 | 10400 |
| chr1_20001_20400 | 34 | 36 | chr1 | 20001 | 20400 |
2. Computing pseudocells¶
Compute metacells is an interesting step to reduce sparsity in our data, since scATAC-seq data have usually a lot of dropouts.
metacells = ci.metacells.compute_metacells(atac)
You can also use your own dimensionality reduction space, that would be stored in the atac.obsm slot
atac.obsm["dim_reduction_name"] = atac.X
ci.metacells.compute_metacells(atac, dim_reduction="dim_reduction_name")
Using adata.obsm['dim_reduction_name'] to identify neighboring cells
AnnData object with n_obs × n_vars = 300 × 1568
var: 'n_cells', 'n_counts', 'chromosome', 'start', 'end'
3. Calculating co-accessibility¶
We can finally compute all the cis co-accessibility scores !
The default way is to indicate your organism if among the one known by Circe.
The atac network is stored automatically as a sparse matrix in atac.varp["atac_network"]
ci.compute_atac_network(
metacells,
njobs=3, verbose=0,
organism="human"
)
Calculating co-accessibility scores...
Concatenating results from all chromosomes...
4. Extract connections¶
4.A.Get the whole genome cis-coaccessible network¶
You can extract connections from the atac.varp slot (sparse matrix), as a DataFrame object with extract_atac_links.
circe_network = ci.extract_atac_links(metacells) #atac)
circe_network.head(3)
| Peak1 | Peak2 | score | |
|---|---|---|---|
| 0 | chr1_1480001_1480400 | chr1_1540001_1540400 | 0.377099 |
| 1 | chr4_1600001_1600400 | chr4_1680001_1680400 | 0.323789 |
| 2 | chr4_10001_10400 | chr4_90001_90400 | 0.298156 |
4.B. Subset the AnnData object to a given window¶
If you’re interested in a specific genomic region only, you can also subset your anndata object on this specific window
subset_atac = ci.subset_region(metacells, "chr1", 10_000, 200_000).copy()
circe_network_subset = ci.extract_atac_links(subset_atac)
circe_network_subset.head(3)
| Peak1 | Peak2 | score | |
|---|---|---|---|
| 0 | chr1_70001_70400 | chr1_130001_130400 | 0.092886 |
| 1 | chr1_60001_60400 | chr1_90001_90400 | 0.000373 |
| 2 | chr1_100001_100400 | chr1_190001_190400 | 0.000067 |
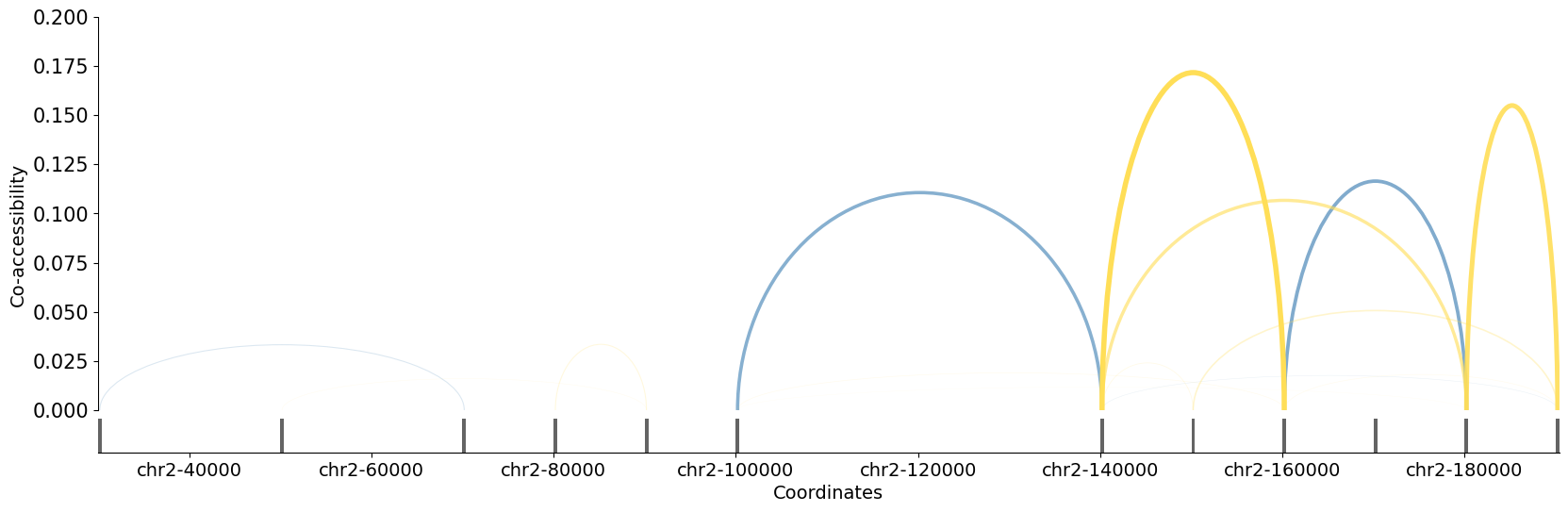
5. Plot co-accessibility scores in a window¶
You can pass either your anndata object or your freshly extracted dataframe into plot_connections to visualize all Circe scores.
Blue edges correspond to positive co-accessibility scores, while yellow ones are for negative scores
ci.plot_connections(
circe_network,
chromosome="chr2",
start=10_000,
end=200_000,
sep=("_","_"),
abs_threshold=0.01
)
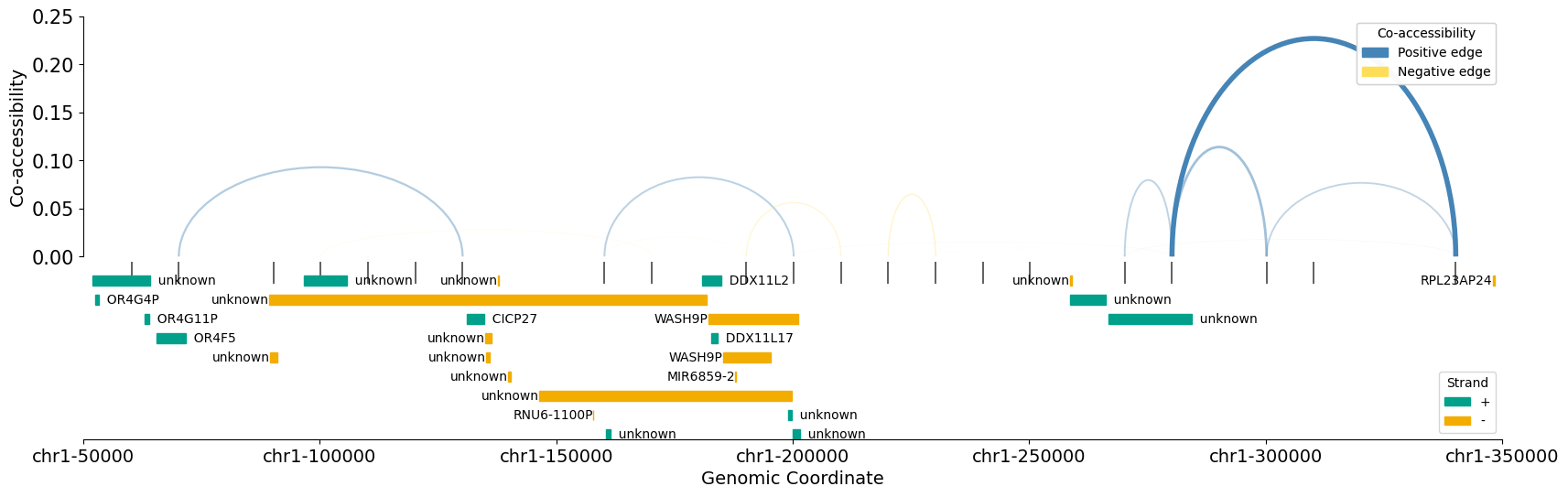
7.Coordinates overlap between co-accessible regions and gene bodies¶
To better understand the role of specific regions and modules, you can additionally plot gene bodies falling into your window of interest.
You need first to load gene coordinates as a dataframe, or to download them through circe.downloads.download_genes. This functions will require to install the pybiomart package Then you can use ci.draw.plot_connections_genes, using gene infos and either your AnnData object or the dataframe of Circe’s results to compare these locations.
pip install pybiomart
Requirement already satisfied: pybiomart in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (0.2.0)
Requirement already satisfied: future in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pybiomart) (1.0.0)
Requirement already satisfied: pandas in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pybiomart) (2.2.3)
Requirement already satisfied: requests in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pybiomart) (2.32.3)
Requirement already satisfied: requests-cache in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pybiomart) (1.2.1)
Requirement already satisfied: numpy>=1.22.4 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pandas->pybiomart) (1.26.4)
Requirement already satisfied: python-dateutil>=2.8.2 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pandas->pybiomart) (2.9.0.post0)
Requirement already satisfied: pytz>=2020.1 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pandas->pybiomart) (2025.1)
Requirement already satisfied: tzdata>=2022.7 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from pandas->pybiomart) (2025.1)
Requirement already satisfied: charset_normalizer<4,>=2 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests->pybiomart) (3.4.1)
Requirement already satisfied: idna<4,>=2.5 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests->pybiomart) (3.10)
Requirement already satisfied: urllib3<3,>=1.21.1 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests->pybiomart) (2.2.1)
Requirement already satisfied: certifi>=2017.4.17 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests->pybiomart) (2024.12.14)
Requirement already satisfied: attrs>=21.2 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests-cache->pybiomart) (25.1.0)
Requirement already satisfied: cattrs>=22.2 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests-cache->pybiomart) (24.1.2)
Requirement already satisfied: platformdirs>=2.5 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests-cache->pybiomart) (4.3.6)
Requirement already satisfied: url-normalize>=1.4 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from requests-cache->pybiomart) (1.4.3)
Requirement already satisfied: exceptiongroup>=1.1.1 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from cattrs>=22.2->requests-cache->pybiomart) (1.2.2)
Requirement already satisfied: typing-extensions!=4.6.3,>=4.1.0 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from cattrs>=22.2->requests-cache->pybiomart) (4.12.2)
Requirement already satisfied: six>=1.5 in /home/rtrimbou/miniconda3/envs/circe_local/lib/python3.10/site-packages (from python-dateutil>=2.8.2->pandas->pybiomart) (1.17.0)
Note: you may need to restart the kernel to use updated packages.
import circe.downloads
genes_df = ci.downloads.download_genes()
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1, figsize = (20, 6))
ci.draw.plot_connections_genes(
connections=metacells,
genes=genes_df,
chromosome="chr1",
start=50_000,
end=350_000,
gene_spacing=30_000,
y_lim_top=-0.01,
abs_threshold=0.0,
track_spacing=0.01,
track_width=0.01,
legend=True,
ax=ax
)
7. Work in progress! Happy to get feedbacks :)¶
If you feel any function could be useful for you on others, don’t hesitate to write me at remi.trimbour@pasteur.fr or to submit an issue on github.com/cantinilab/Circe.